
Research Interests/Area of Study: Molecular dissection and translational research of the p53 and c-myc networks in controlling metabolism, cell growth, senescence, death, differentiation, aging, and tumorigenesis.
Summary: The Lu laboratory is interested in understanding the molecular and biochemical basis that underlies physiological and pathological signaling pathways (growth, metabolic, hypoxia, or DNA damage signals), which lead to gene expression and subsequent cell growth arrest, differentiation, senescence, autophagy, or apoptosis. Read More
The abnormal alterations of these pathways often result in and/or facilitate tumorigenesis. A remarkable example of the tumorigenic abnormality is the alternation of the components in the stress signaling pathway that is mediated by the p53 tumor suppressor protein and its negative regulators, such as MDM2 and MDMX. Genetic studies show that MDM2 and MDMX are the physiological feedback regulators of p53, and these proteins play important roles in tumorigenesis. MDM2 and MDMX repress p53 function by mediating its degradation and directly suppressing its activity. Decetylation of p53 mediated by SIRT1 or HDAC1 can facilitate MDM2/MDMX-mediated p53 suppression. Various stress signals lead to p53 activation by blocking this feedback regulation. Recently, my laboratory has identified a novel class of small molecules, named Inauhzin that can inhibit SIRT1 and IMPDH2 activity, activate p53 and induce p53-dependent apoptosis and senescence, consequently suppressing tumor growth. Another example of the cancerous abnormality is the overexpression of oncogenes, such as c-myc or mutated p53s. Our recent studies show that ribosomal proteins regulate c-Myc activity. Also, a newly identified miRNA can regulate c-Myc expression and activity in an auto-regulatory fashion. To understand the molecular and biochemical mechanisms for cell proliferation and tumorigenesis involving the p53 and c-Myc pathways, my laboratory focuses on the following projects:
- To understand the biochemical mechanism underlying the regulation of MDM2 by ribosomal proteins, leading p53 activation, and the role of these ribosomal proteins and their regulators, such as RBM10 or Spin1, in cell cycle regulation and tumorigenesis;
- To elucidate molecular mechanisms for the AMPK regulation of MDMX, leading p53 activation, in response to metabolic and hypoxia signals by using genetically manipulated mouse models, such as a double knockin (DKI) mouse line that harbors both MDM2 and MDMX point mutation, impairing the ribosomal stress-MDM2- and metabolic stress-AMPK-MDMX-p53 pathways;
- To illustrate the roles of several newly identified p53 targets, such as PHLDB2, NGFR, or GRIN2C, in controlling p53-dependent and p53-independent functions during tumorigenesis and drug resistance;
- To determine the roles of p53 hot spot mutations in tumor development, progression and drug resistance, such as p53 R249S in HCC, or R273H and R248W in lung and colon cancer metabolism, development and drug resistance, particularly revealing the molecular insights into the roles of their GOFs in cancer stem cell proliferations and cancer development as well as cancer immunology;
- To determine the role of CCDC3, a newly identified p63 target, in lipid metabolism, fatty liver development, and inflammatory response as well as the association of these physiological and pathological events with cancer development and progression;
- To understand the role of unique posttranslational modifications of p53 or c-Myc in regulation of these transcriptional factors’ functions and cancer biology;
- To ultimately develop anti-cancer drugs by targeting the p53 (such as Inauhzin) and c-Myc pathways.
Diverse approaches including quantitative and analytical protein biochemistry, chemical biology, proteomics, immunological tools, gene microarray, RNA seq, molecular and cellular biological methods as well as genetic methods (such as murine model, orthotopic and PDX tumor model systems) will be employed in these studies. We will also pursue translational research by screening anti-cancer drugs targeting the above pathways and examining molecular alternations of these pathways in human cancers. The effort will be complemented by collaborating with other groups on and off the campus.
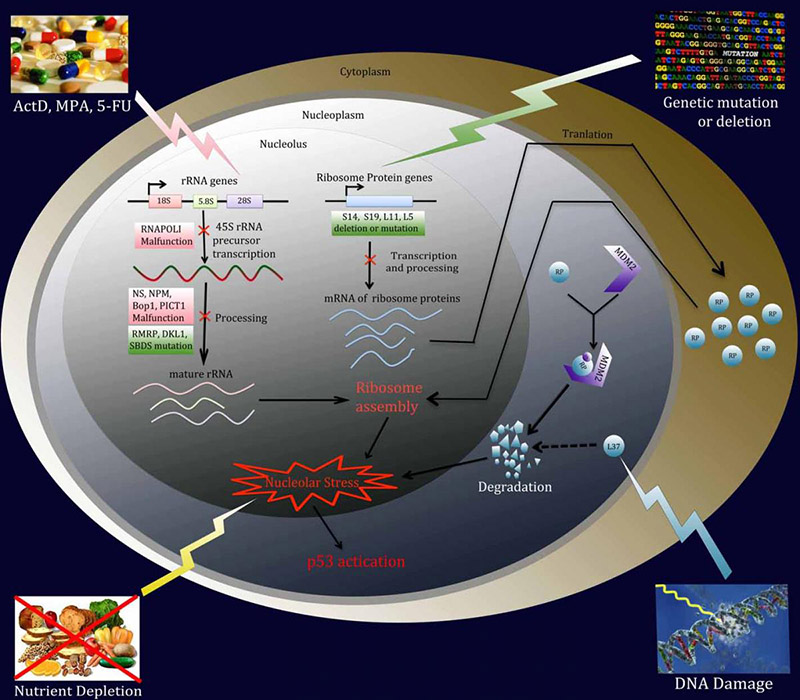
Figure 1. A variety of environmental reagents and genetic alterations can lead to nucleolar or ribosomal stress. Ribosome biogenesis involves 3 major events: (1) synthesis and processing of rRNA, (2) synthesis of RPs, and (3) assembly and cellular transportation of 40S and 60S ribosome units. Perturbation of each of these steps triggers nucleolar or ribosomal stress.
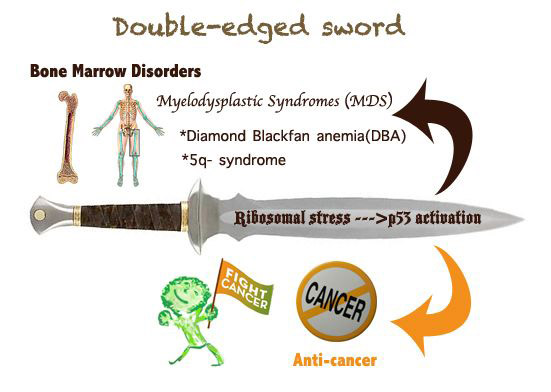
Figure 2. A double-edged sword: the nucleolar stress-p53 activation. The RP-mediated p53 activation by inactivating MDM2 upon nucleolar stress causes a dual effect on human health. On one hand, p53 activation prevents or retards tumor growth in response to nucleolar stress; on the other hand, inappropriate p53 activation due to ribosome dysfunction leads to myelodysplastic syndromes (MDS), such as DBA or 5q syndrome.
